Introduction
Isoform phasing and quantification is one of the major objectives and challenges in transcriptome studies. Second-generation sequencing (SGS) made it possible to evaluate the expression and level of transcribed genes. However, the SGS reads have limited length, causing the difficulty both in capture of full-length transcripts and in resolving the linkage of SNPs (isoform phasing). In recent years, the advance of single-molecule sequencing technology has extended the length of reads significantly, complementing SGS in recognition of complete isoforms at single-molecule resolution effectively.
However, the SMS also has its own problems. The most outstanding drawbacks include (1) high sequencing errors, and (2) relatively low throughput. The low throughput and inherently large variation of isoform expression further derived the third problem – the effective number and variety of isoforms captured by SMS would be very limited. Sequencing errors could be partly corrected by increase of coverage depth of the molecules and read alignment based consensus sequence analysis. However, the other two problems and high computational requirements for the indel-prone long read alignment constrained the above self-correction. SMS error correction by parallel SGS reads could be an ideal resolution, since the SGS reads are highly accurate while the throughput and cost are not either a problem. Both the two main SMS techniques, PacBio and Nanopore, have continued to release new platforms that could hopefully increase the throughput and reduce the costs. Other strategies were also proposed, e.g., targeted sequencing and cDNA normalization, which could also improve the isoform variety problem caused by low throughput and uneven expression of transcripts.
A few software tools have been developed to detect the isoforms from SMS sequences corrected by SGS reads (e.g., IDP), to phase isoforms purely based on SMS reads (e.g., HapIso) or using both SMS and SGS data (e.g., IDP-ASE). IDP-ASE can also quantify the isoforms phased. Here, we proposed an alternative but simpler method, ASIIQT, to correct SMS sequences, phase isoforms and quantify the allele-specific isoforms simultaneously. Not like HapIso, ASIIQT uses parallel SGS reads to predefine SNPs and reduce noises caused by the high SMS sequencing errors. HapIso also misses all indels while ASIIQT does not. Compared to IDP-ASE, ASIIQT mainly depends on SMS reads rather than SGS reads to phase the isoforms. The smaller contribution of SMS data in isoform phasing for IDP-ASE was likely due to the small variety of detected isoform species by original SMS. We found that the cDNA normalization could well improve the diversity of isoforms detected by SMS and therefore the SMS data could provide much more guidance for isoform phasing. ASIIQT could also correct and quantify the isoforms with SGS reads.
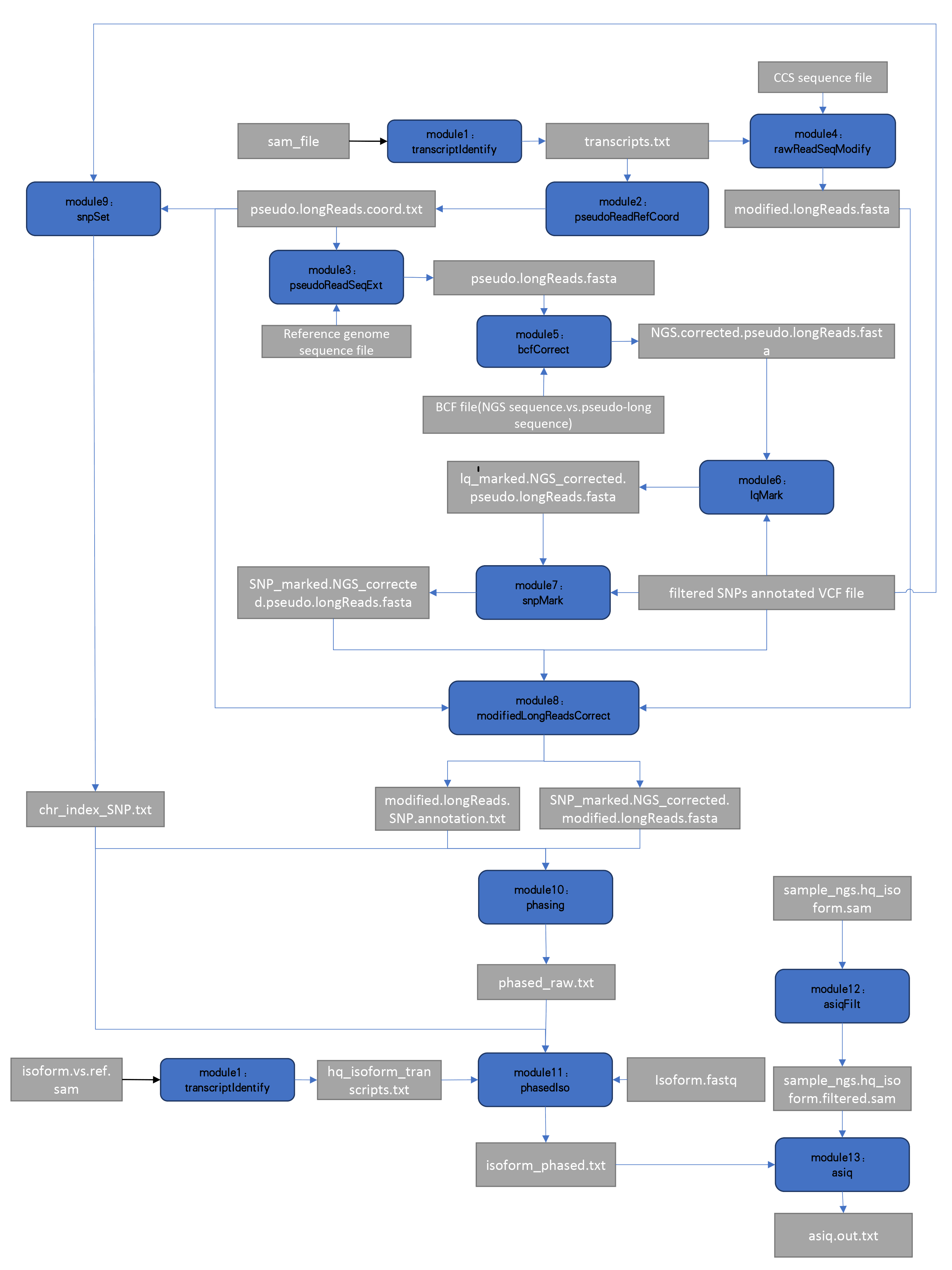
The ASIIQT pipeline is straightforward. Simply, for each cDNA library, both cDNA-normalized SMS and direct SGS are performed. SMS reads are mapped to genome references and pseudo-long reads are generated, for which the SMS read sequences are replaced by low-error reference fragmental sequence. The SGS reads could be mapped to the pseudo-long reads efficiently by DBG algorithms because neither the latter sequences have many indel mutations nor they contain introns, both of which would influence the mapping process. Using the pseudo-long reads as bridges, the original SMS reads could be corrected easily. Before correction, SGS reads are also mapped to reference genome and the SNP set is identified. The SNP loci within SMS reads are not modified during the SMS read correction process. The semi-corrected SMS sequences are collected and used for SNP linkage analysis. After phasing, the NGS reads are mapped to the corrected isoforms and for each NGS read at most two hits are allowed to be output, the uniquely-mapped reads for each isoform are collected, and the phased SNPs are distinguished and quantified.